
Imagine spending millions developing a new drug, only to have it rejected because it degraded during shipping. That is the harsh reality of pharmaceutical development without proper stability testing. This process isn't just paperwork; it is the scientific proof that your medicine remains safe, effective, and potent until the patient uses it. For anyone working in bioequivalence or regulatory affairs, understanding the exact stability testing requirements regarding temperature and time is non-negotiable. One wrong setting in a climate chamber can invalidate months of data and delay market approval by years.
The global standard for this work comes from the International Council for Harmonisation (ICH). Specifically, guideline ICH Q1A(R2) dictates how we test drug substances and finished products. It harmonizes rules across the US FDA, European Medicines Agency (EMA), and Health Canada. If you follow these guidelines, you avoid redundant testing and ensure your product meets international safety standards. But the devil is in the details-specifically, the precise combinations of heat, humidity, and duration required for different climatic zones.
Core Stability Testing Conditions: The ICH Framework
To establish a valid shelf life, you must run three types of studies: long-term, accelerated, and intermediate. Each has strict temperature and relative humidity (RH) parameters defined by ICH Q1A(R2). These are not suggestions; they are regulatory mandates.
| Study Type | Temperature | Relative Humidity (RH) | Duration at Submission |
|---|---|---|---|
| Long-Term Study | 25°C ± 2°C OR 30°C ± 2°C | 60% RH ± 5% OR 65% RH ± 5% | 12 months (FDA) / 6-12 months (EMA) |
| Accelerated Study | 40°C ± 2°C | 75% RH ± 5% | 6 months |
| Intermediate Study | 30°C ± 2°C | 65% RH ± 5% | Required if significant change occurs in accelerated study |
For long-term studies, you choose between 25°C/60% RH and 30°C/65% RH based on where you plan to sell the drug. If your target market is temperate (like Northern Europe or North America), 25°C/60% RH is standard. For hotter, more humid regions (like Southeast Asia or Africa), 30°C/65% RH is often required. The FDA strictly requires 12 months of long-term data before you can submit an application. The EMA offers some flexibility, allowing either 6 or 12 months depending on the submission option, which can speed up approvals in Europe but complicate global launches.
Accelerated testing at 40°C/75% RH serves as a stress test. It simulates extreme conditions like a package left in a hot car or a warehouse with AC failure. Dr. John B. Sullivan, former FDA Division Director, noted that this specific temperature was chosen to represent extreme shipping excursions while staying below the melting point of most excipients. If your drug degrades significantly here, you must trigger an intermediate study at 30°C/65% RH to better predict real-world performance.
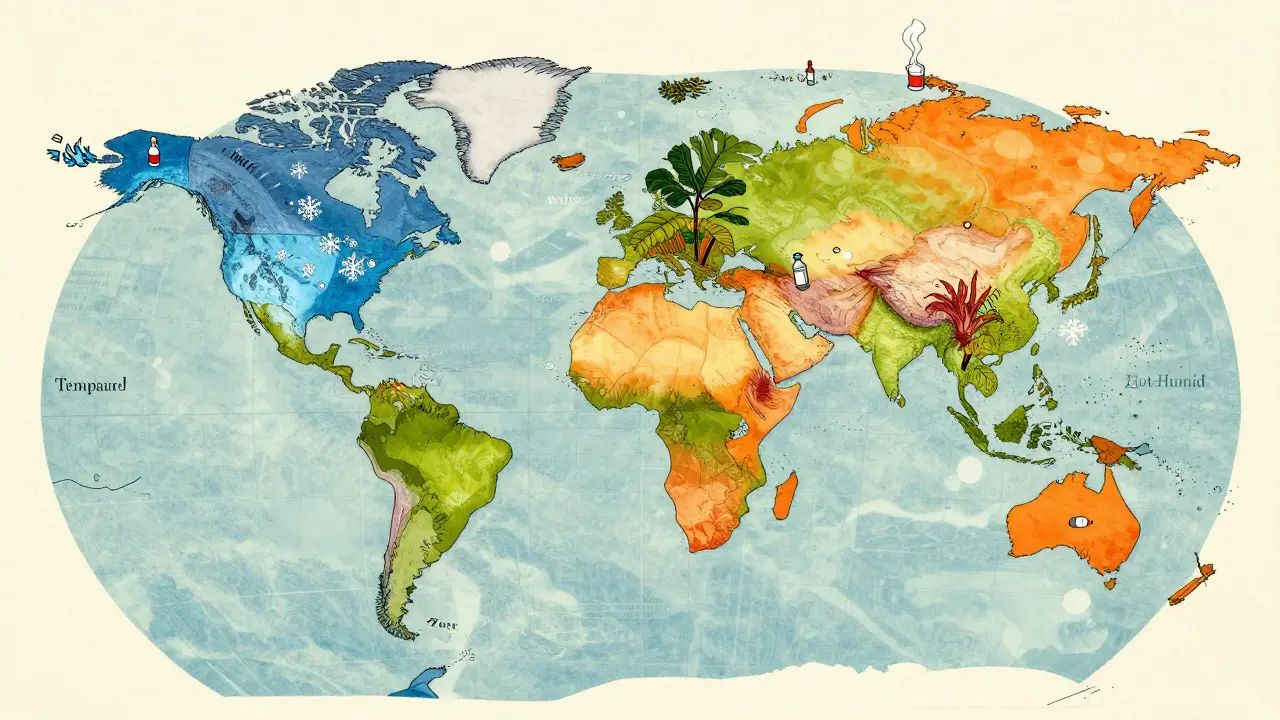
Climatic Zones and Regional Variations
Not all parts of the world share the same weather patterns, so the World Health Organization (WHO) and ICH define five climatic zones. Your stability protocol must match the zone where the drug will be stored and used.
- Zone I (Temperate): 21°C / 45% RH. Examples include Canada and Northern Europe.
- Zone II (Subtropical/Mediterranean): 25°C / 60% RH. Examples include Southern Europe and parts of the USA.
- Zone III (Hot-Dry): 30°C / 35% RH. Examples include the Middle East.
- Zone IVa (Hot-Humid/Tropical): 30°C / 65% RH. Examples include India and Southeast Asia.
- Zone IVb (Hot-Higher Humidity): 30°C / 75% RH. Examples include parts of Africa and the Caribbean.
If you are targeting Zone IV markets, you cannot simply rely on standard 25°C/60% RH data. You need separate protocols that account for higher baseline temperatures and humidity. This adds complexity and cost. According to a 2023 industry survey by Tovatech, companies targeting Zone IV markets face 4-6 month delays in development timelines due to these additional stability protocols. Ignoring these zones can lead to product recalls once the drug hits the market and fails under local storage conditions.
Bioequivalence and Stability: The Critical Link
When discussing generic drugs, bioequivalence (BE) is the gold standard. But BE data is only valid if the drug substance remains stable. A common pitfall is assuming that because two generics have identical chemical structures, their stability profiles are interchangeable. They are not.
Excipients-the inactive ingredients like binders, fillers, and coatings-play a huge role in stability. Two generic versions of the same API might use different polymers or lubricants. Under high humidity, one polymer might absorb moisture and swell, altering the dissolution rate. This changes the bioavailability, breaking the bioequivalence even if the assay purity looks fine.
Regulators require that the stability-indicating methods used in your BE study are validated. If your HPLC method cannot distinguish between the main drug peak and its degradation products, your stability data is useless. The FDA’s 2022 warning letters frequently cited inadequate stability-indicating methods as a primary reason for rejection. Ensure your analytical methods are robust enough to detect hydrolysis, oxidation, and photodegradation early in the timeline.
Refrigerated Products and Special Cases
Biologics, vaccines, and certain injectables require cold chain management. Their stability requirements differ significantly from small-molecule tablets. For refrigerated products, the long-term condition is typically 5°C ± 3°C. There is no humidity requirement for frozen products, but for refrigerated liquids, condensation can be a risk.
Accelerated testing for refrigerated items does NOT use 40°C. Instead, WHO guidelines specify 25°C ± 2°C / 60% RH ± 5% RH for 6 months. Using 40°C on a protein-based vaccine would denature the molecule instantly, providing no useful predictive data. The FDA allows some flexibility here, permitting 30°C or even higher humidity levels based on a risk assessment, but you must justify this deviation scientifically. Misapplying ambient conditions to cold-chain products is a frequent error among junior analysts.

Practical Implementation: Avoiding Common Pitfalls
Even with perfect protocols, execution errors can ruin your data. Here are the most common issues professionals face:
- Chamber Calibration Failures: ISO 14644-1 standards require chambers to maintain temperature within ±0.5°C and humidity within ±2% RH. In practice, many labs struggle with this. A 2023 LinkedIn survey of stability professionals found that 78% experienced at least one temperature excursion exceeding ±2°C during a 12-month study. Always perform IQ/OQ/PQ (Installation, Operational, Performance Qualification) before starting any study.
- Subjective "Significant Change": ICH defines significant change as a shift outside specification limits. However, regulators sometimes reject batches that are technically within spec but show a worrying trend. For example, a Pfizer employee reported a case where a 4.8% drop in assay (still within 95-105%) triggered regulatory scrutiny. Document every minor variation. Statistical trends matter as much as absolute values.
- Packaging Interactions: The container closure system interacts with the drug. Blister packs, vials, and bottles offer different barriers to oxygen and moisture. Test your final packaging configuration, not just bulk powder. Merck’s success with Keytruda® involved using intermediate conditions to detect a polymorphic transition that only occurred in specific blister pack materials.
Documentation is your defense. An average stability dossier contains 450-600 pages. Include raw data, environmental logs, and annual reports. If you outsource to a CRO (Contract Research Organization), verify their chamber mapping data. Temperature variations across shelves can reach ±1.8°C in poorly mapped large chambers. Place your samples in the center of the chamber, away from doors and fans, to ensure uniform exposure.
Future Trends: Beyond Static Conditions
The landscape is shifting. The traditional ICH Q1A(R2) framework, unchanged for over two decades, is under review. The ICH Working Group is proposing updates to address complex products like antibody-drug conjugates (ADCs) and cell therapies. These biologics degrade via mechanisms not captured by static heat and humidity tests, such as freeze-thaw cycles or shear stress.
Real-time release testing (RRT) and Process Analytical Technology (PAT) are gaining traction. The FDA’s 2023 pilot program suggests that continuous manufacturing could reduce stability testing duration by 30-50%. Predictive modeling using AI is also emerging. Companies like WuXi AppTec are implementing Accelerated Predictive Stability (APS) studies at extreme temperatures (50-80°C) to model degradation pathways faster. While regulators remain cautious-EMA rejected several model-based submissions in 2022-the trend toward risk-based, dynamic testing is inevitable.
What is the difference between long-term and accelerated stability testing?
Long-term testing mimics real-world storage conditions (e.g., 25°C/60% RH) to determine the actual shelf life. Accelerated testing (40°C/75% RH) stresses the product to identify potential degradation products and estimate stability under abusive conditions. Accelerated data helps predict long-term behavior but does not replace real-time data for labeling claims.
How long does stability testing take before submission?
The FDA requires 12 months of long-term stability data at the time of submission for new drug applications. The EMA may accept 6 months of data under certain options. Accelerated testing always runs for 6 months. Total project timelines often extend to 12-24 months to cover all time points (0, 3, 6, 9, 12 months).
Why is humidity control critical in stability testing?
Humidity drives hydrolysis and affects hygroscopic compounds. Many solid oral dosage forms fail due to moisture absorption, which can alter dissolution rates and bioavailability. The AAPS notes that 62% of stability failures in solids result from humidity cycling. Precise control (±5% RH) is essential for accurate data.
Can I use accelerated data to set the expiration date?
No. Accelerated data alone cannot support a labeled expiration date. It is used to propose tentative dates and identify degradation pathways. Only long-term real-time data can confirm the final shelf life approved by regulators. However, if no significant change occurs in accelerated studies, it supports the proposed long-term shelf life.
What happens if my product fails stability testing?
If a product fails, you must investigate the root cause. Options include reformulating the drug, changing the packaging to provide better barrier protection, or adjusting storage instructions (e.g., requiring refrigeration). You cannot simply ignore the failure. Regulatory authorities will reject applications with unexplained stability failures.














Write a comment
Your email address will be restricted to us